 Maldição de Ondina (Hipoventilação alveolar primária) Frequência: Entre 200-300 casos conhecidos em todo mundo. Por ser causa de morte súbita pensa-se que os casos conhecidos são só a ponta do iceberg e que na realidade 1 bebé a cada 200.000 nasçam com esta anomalia. Causa: Parcialmente conhecida. A principal causa é uma mutação ou vários do gene PHOX2B, de herança autossômica dominante. Os mecanismos da respiração involuntária não funcionam adequadamente. Ao dormir, os receptores químicos que recebem sinais (baixa de oxigênio ou aumento de dióxido de carbono no sangue) não chegam a transmitir os sinais nervosos necessárias para que se dê a respiração.
Maldição de Ondina (Hipoventilação alveolar primária) Frequência: Entre 200-300 casos conhecidos em todo mundo. Por ser causa de morte súbita pensa-se que os casos conhecidos são só a ponta do iceberg e que na realidade 1 bebé a cada 200.000 nasçam com esta anomalia. Causa: Parcialmente conhecida. A principal causa é uma mutação ou vários do gene PHOX2B, de herança autossômica dominante. Os mecanismos da respiração involuntária não funcionam adequadamente. Ao dormir, os receptores químicos que recebem sinais (baixa de oxigênio ou aumento de dióxido de carbono no sangue) não chegam a transmitir os sinais nervosos necessárias para que se dê a respiração.
Descrição: Nas formas mais leves da maldição de Ondina, o sujeito poderá viver normalmente, mas estará sempre sonolento durante o dia, se cansará facilmente, constante dor de cabeça com aumento do nível de glóbulos vermelhos. A Síndrome de Ondine (Hipoventilação alveolar primária) é uma doença na qual a principal causa é uma mutação ou várias do gene PHOX2B, de herança autossômica dominante. Nessa doença os mecanismos da respiração involuntária não funcionam adequadamente. Ao dormir, os receptores químicos que recebem sinais (baixa de oxigênio ou aumento de dióxido de carbono no sangue) não chegam a transmitir os sinais nervosos necessários para que se dê a respiração. O nome é uma referência a Ondina, ninfa das águas na mitologia germânica.
Em 2006 havia apenas 200 casos conhecidos em todo o mundo, mas na realidade estima-se que o número de casos seja muito maior (cerca de 1 bebê a cada 200.000 nascimentos), por ser causa de morte de morte súbita, a maioria dos bebês morrem sem que muitas vezes saiba da causa.Na maioria das vezes a síndrome é de leve a moderada, ou seja,a pessoa poderá viver normalmente, mas na maioria dos casos vai precisar de suporte ventilação mecânica para dormir.Nas formas mais graves a pessoa precisa de ventilação mecânica 24 horas por dia, nesses casos é indicado uma cirurgia para colocação de um marca-passo no nervo frênico, a cirurgia e o aparelho (MARK IV) tem um alto custo e mesmo nos países de primeiro mundo são poucos os hospitais que a realizam.
Nas formas mais graves costuma aparecer desde o nascimento, e a maioria de bebês morrem sem que muitas vezes se chegue a saber a causa. No entanto, naquelas pessoas em que a doença piora progressivamente e podem causar a morte de quem dorme, costuma se tratar com ventilação assistida durante a noite.
Síndrome de Proteus

Frequência: 200 casos documentados em todo mundo atualmente. Estima-se que surja um caso por mais de um milhão de nascimentos. Alguns médicos espeialistas defendem que provavelmente seja causado por um gene dominante letal. Outros dizem que se deva a uma recombinação no embrião dando lugar a três tipos de células: Células normais, células de crescimento mínimo e células de crescimento excessivo.
Descrição: Existem uma grande quantidade de malformações cutâneas e subcutâneas, com hiperpigmentação, malformações vasculares e crescimento irregular dos ossos. Produz-se o gigantismo parcial dos membros ou o crescimento excessivo dos dedos enquanto algumas zonas do corpo crescem menos do que deveriam. Tudo isto provoca uma desfiguração extrema da pessoa que costuma ser socialmente estigmatizada. Josep Merrick, o famoso "Homem Elefante", sofria desta síndrome.
A síndrome de Proteus é uma doença congênita que causa crescimento exagerado e patológico da pele com tumores subcutâneos, desenvolvimento atípico com macrodactilia e hemi-hipertrofia. É uma doença extremamente rara: se descreveram cerca de 100 casos em todo mundo. Por causa dessa raridade, não há muitos estudos na área atualmente, e quase todos os acontecimentos, praticamente, não têm solução.
Esta doença teria permanecido desconhecida, se não fosse o caso de Joseph Merrick, O homem elefante, foi um caso particularmente grave desta síndrome, além da neurofibromatose que também acredita-se que tinha.
Em 1980 foi rodado o filme "O Homem Elefante" (The Elephant Man) dirigido por David Lynch e estrelado por Anthony Hopkins, John Hurt, Anne Bancroft e John Gielgud. No filme, o ator John Hurt interpreta Joseph Carey Merrick, onde é relatada sua vida e as dificuldades enfrentada por ele portar a doença que provocou deformidades em 90% do seu corpo.
Em vida, Joseph, quando criança foi rejeitado por sua aparência, sendo assim, expulso de casa. Após isso, tentou vender livros, mas não obteve muito sucesso por causa de sua aparência. Depois de meses tentando arrecadar fundos para viver, foi "resgatado" por um circo de aberrações, onde foi posto como atração sob o título de "A parte mais degradante do ser humano". Após tempos de circenses, um médico dos arredores o recolheu para análise e tratamento. Em 1890, Joseph veio a falecer por causa do crescimento de seu cérebro e seu rompimento de pele.
Síndrome Hutchinson-Gilford
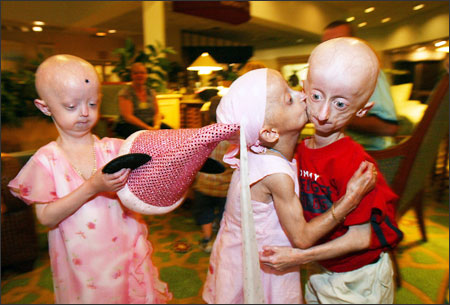
Frequência: Ao redor de 100 casos documentados. Estima-se que aparece um caso da doença a cada 8 milhões de nascimentos, ainda que poderia ser maior já que muitas vezes não chega a ser diagnosticada.
Causa: Parcialmente conhecida. A maioria dos casos são produzidas por mutações de herança autossômica dominante no gene LMNA. Este gene participa na manutenção da estabilidade nuclear e a organização da cromatina. Não há cura, e até a conclusão da pesquisa, no final do ano passado, sua causa era desconhecida. Agora, cientistas franceses e norte-americanos descobriram que a doença está ligada a um gene que controla a estrutura do núcleo das células. Neste caso as pesquisas estão avançando para uma provável cura da doença.
Descrição: Os indivíduos com esta síndrome envelhecem muito rapidamente desde a infância. No nascimento têm uma aparência totalmente normal, mas crescem cada vez mais lentamente que as outras crianças e desenvolvem uma expressão facial muito característica. Perdem o cabelo, adquirem rugas e padecem de um dano severo das artérias (arteriosclerose) que causa à morte nos primeiros anos da adolescência. É uma doença genética extremamente rara que acelera o processo de envelhecimento em cerca de sete vezes em relação à taxa normal. Uma criança com 10 anos se parece com uma pessoa de 70 anos. A palavra progeria é derivada do grego e significa "prematuramente velho". A expectativa média de vida das pessoas é de 14 anos para as meninas e 16 para os meninos. Desde a sua identificação foram relatados cerca de 100 casos. As vítimas de progeria nascem bebês normais mas, por volta dos 18 meses, começam a desenvolver sintomas de envelhecimento precoce.
Rabo Humano (Rabo Vestigial)

Frequência: Ao redor de 100 casos documentados em todo mundo.
Causa: Não se conhece em profundidade. Acha-se que é produzido pela mutação dos genes encarregados de produzir a morte celular das células que estavam destinadas a formar um rabo.
Descrição: Observa-se a presença de uma rabo na zona final do sacro, no nível do cóccix. O rabo é composto de músculos, vasos sanguíneos, nervos, pele, vértebras e cartilagem. Embora seja bastante rara, com cerca de 100 casos documentados na literatura da medicina, existe também essa anomalia. Trata-se de bebês que nascem com uma cauda (rabinho mesmo) bem desenvolvido, formado ainda durante a gestação.
Mas como isso acontece? Na verdade, não se sabe ao certo, mas sabemos que nossos ancestrais hominídeos tinham rabo e com a evolução esta estrutura foi perdida e isso aconteceu bem antes de se tornarem bípedes (andam em duas patas, ou pés). Hoje em dia temos genes que são encarregados de produzir a morte celular das células que estavam destinadas a formar o rabo e isso já vem acontecendo a milhares de anos. Acredita-se que de alguma forma, ocorre nessas pessoas uma mutação destes genes e aquelas células não são destruídas e acabam formando o rabinho!
No entanto, há dois tipos de rabo humano: o falso rabo e o rabo humano verdadeiro, muito mais raro. O falso rabo não tem ossos nem cartilagem, é pele e gordura. Porém, o rabo humano verdadeiro tem nervos e músculos e, às vezes, até cartilagens ou vértebras. Neste caso, o rabinho possui uma musculatura associada as vértebras que permite seu movimento.
Gêmeo Parasita (Fetus in Fetus)
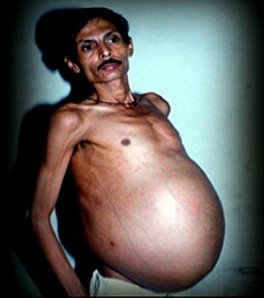
Frequência: Ao redor de 100 casos documentados em todo mundo.
Causa: É um exacerbo do caso dos siameses. Dois gêmeos não chegam a se separar completamente quando são zigotos e ficam unidos por alguma zona. Um destes gêmeos cresce enquanto o outro se atrofia ficando no interior do gêmeo são e dependendo completamente dele. Desconhece-se por que os gêmeos não se separam corretamente.
Descrição: Quando o feto hospedador consegue sobreviver ao parto, este fica com um inchaço na zona onde se situe o feto parasita. 80% das vezes encontra-se na região abdominal, mas também pode se encontrar no crâneo e até no escroto. Também pode passar desapercebido no princípio. Mais tarde, conforme a pessoa vai crescendo também cresce o feto parasita.
Ao realizar provas de imagem observam-se órgãos em lugares onde não deveriam existir ainda que também podem se ver umas diminutas pernas, braços, dedos, cabelo ou qualquer outro elemento do feto que tenha desenvolvido. Não há dois casos iguais de "fetus in fetus", já que os fetos parasitas podem se situar em zonas muito diferentes do feto hospedador e, por tanto, também será diferente o grau de crescimento e elementos que tenha chegado a desenvolver. Há fetos parasitas muito desenvolvidos e outros que só possuem um número escasso de órgãos.
Esta anomalia poderia ser dizer que é um exacerbo do caso dos gêmeos xipófagos (siameses). Neste caso, é considerada uma anomalia “fetus in fetu“, quando um gêmeo malformado é encontrado dentro do corpo de um gêmeo hospedeiro, seja uma criança ou um adulto vivo.
Isso mesmo, os gêmeos não chegam a se separar completamente quando são zigotos e ficam unidos por alguma região do corpo. Um destes gêmeos cresce, se desenvolve “normalmente”, enquanto o outro se atrofia e se aloja no interior do gêmeo sadio e passa a depender completamente dele.
De acordo com a literatura especializada, quando o feto hospedeiro consegue sobreviver ao parto, este fica com uma protuberância, como se fosse um inchaço na região onde está feto parasita. Cerca de 80% dos casos ocorrem com o parasita alojado na região abdominal. Quando nasce uma criança (ou um adulto passa por exames específicos), podem ser escontradas no abdomen massas contendo ossos, cartilagem, dentes, tecido do sistema nervoso central, gordura e músculo denominadas ‘teratomas’. Mas só serão consideradas como fetus in fetu caso haja um tronco e membros reconhecidos.
Embora seja difícil saber a taxa exata de incidência, já que os casos podem passar despercebidos durante longos períodos, acredita-se que o fetus in fetu ocorre a cada grupo de 500 mil nascimentos, e muitas vezes passam desapercebidos sem serem diagnosticados.
Síndrome do Homem Lobo (Hipertricose Lanuginosa Congênita)
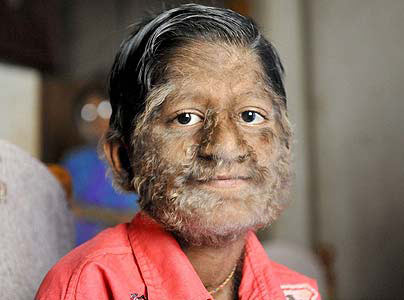
Frequência: 40-50 Casos documentados em todo mundo desde sua descoberta. A incidência natural (sem contar os casos em famílias) estima-se um caso entre 1 a 10 bilhões de habitantes.
Causa: Desconhecida. Pensa-se que é uma mutação e a maioria é de herança familiar e, muito raramente, a mutação dá-se de forma espontânea.
Descrição: As pessoas que padecem da doença ficam completamente cobertas por um longa lanugem (cabelo) excepto nas palmas das mãos e dos pés. O comprimento do cabelo pode chegar a 25 centímetros.
É uma anomalia bastante estranha. As pessoas que sofrem com essa doença ficam completamente cobertas por um longa lanugem (cabelo), com excessão das palmas das mãos e dos pés. O comprimento destes pêlos pode chegar a 25 centímetros. Uau!!!!!
A lanugem é um cabelo fino (como se fosse uma penugem) que aparece nos recém nascidos e que desaparece normalmente pouco antes do nascimento, alguns bebês até nascem com um pouco de lanugem e isso é normal! Mas pessoas com hipertricose não perdem essa pelugem e ainda podem desenvolve-la e se tornarem com a incrível aparência de “lobisonem”. Nesta doença os folículos capilares previamente normais de todos os tipos revertem a produção de pêlos com características de lanugem. Pêlo fino e felpudo cresce sobre grande área do corpo, repondo pêlo normal e veloso primário e secundário.
Na Hipertricose congênita, temos nada mais que 50 casos registrados. Neste caso, ainda durante a gravidez, o feto é coberto com a lanugem que deveria cair durante o oitavo mês de gestação, mas continua a crescer. Os pêlos ficam escuros e permanecem após o nascimento. Ainda não se sabe ao certo as causas, porém acredita-se que seja uma condição genética, considerada hereditária ou então pode acontecer apenas devido à uma mutação de genes. Na Hipertricose Adquirida, o crescimento de pêlos se dá após o nascimento desencadeado geralmente por problemas relacionados ao cancer.
Hipertricose é uma condição que realmente não tem uma cura. Os tratamentos incluem apenas técnicas de depilação avançadas com utilização de laser e eletrólise dos fios, porém nenhuma dessas técnicas são realmente satisfatórias.
Fonte: http://www.mdig.com.br/
http://pt.wikipedia.org/
http://www.fiocruz.br/
http://diariodebiologia.com/
http://www.dihitt.com.br/