 2007 - A escolha destas anomalias, entre as milhares de doenças raras que existem, não foi arbitrária. O padrão adotado para a escolha foi a predominância do alto grau de estranheza e da escassa freqüência da doença. Síndrome Riley-Day, insensibilidade congênita à dor - Frequência: 100 casos documentados nos Estados Unidos. Desconhece-se a frequência em outros locais por não ser facilmente diagnosticado por...
2007 - A escolha destas anomalias, entre as milhares de doenças raras que existem, não foi arbitrária. O padrão adotado para a escolha foi a predominância do alto grau de estranheza e da escassa freqüência da doença. Síndrome Riley-Day, insensibilidade congênita à dor - Frequência: 100 casos documentados nos Estados Unidos. Desconhece-se a frequência em outros locais por não ser facilmente diagnosticado por...
passar quase sempre desapercebida. Causa: Descoberta recentemente. Deve-se a uma mutação num gene encarregado da síntese de um tipo de canal de sódio que se encontra principalmente em neurônios encarregados de receber e transmitir o estímulo doloroso. A doença é causada por uma mutação no gene IKBKAO, que se localiza no cromossomo 9. É uma doença de caráter autossômico recessivo, significando que uma pessoa deve herdar uma cópia do gene defeituoso de cada progenitor para desenvolver a condição.
Descrição: São indivíduos totalmente normais no tato e na sensibilidade ao frio, ao calor, pressão e cosquinhas. No entanto, ante qualquer ato que em pessoas normais provocaria dor (como fincar uma agulha) não provoca nenhuma sensação dolorosa. Como consequência disto, costumam morrer mais jovens por traumatismos e lesões ao não sentir nenhum dano. Devem estar sempre sob o cuidado dos olhos quando crianças para que não se machuquem eles mesmos.
Tratamento: Não existe cura para esta doença, e o tratamento consiste na prevenção de acidentes e na amenização dos outros sintomas, o que pode incluir:
Proteção da pessoa contra lesões;
Tratamento da pneumonia aspirativa;
Terapêutica anticonvulsivante, se o paciente apresentar convulsões;
Medicamentos, que incluem lágrimas artificiais, para evitar a secagem dos olhos e medicamentos chamados antieméticos, que servem apara conrolar possíveis vômitos. A hipotensão ortostática (pressão arterial baixa quando em repouso) pode ser operado com um aumento na ingestão de fluidos e sal, cafeína e meias elásticas ao nível da cintura.
A Síndrome de Riley-Day, ou a sigla ICDA é uma desordem do sistema nervoso autônomo que afeta o desenvolvimento e a sobrevivência dos neurônios sensoriais, simpáticos e parassimpático no sistema nervoso autônomo sensorial, resultando variáveis sintomas incluindo: insensibilidade à dor, incapacidade de produzir lágrimas, fraco crescimento, e pressão arterial lábil (hipertensão episódica e hipotensão postural).
É uma doença hereditária autossómica recessiva relacionada com uma mutação no gene IKBKAP do cromossomo 9. O sintoma mais característico é a ausência da sensibilidade à dor. No entanto, existem ainda outros sintomas: deficiências no crescimento, dificuldades em comer, apneia, vómitos, convulsões, hipotonia, escoliose severa, diminuição do sentido do paladar, … Assim, quem possui a doença não sente dores. Elas ficam muito mais sujeitas a sofrer acidentes porque param de registrar qualquer aviso de dano nos tecidos do corpo, como cortes ou queimaduras. Foi descrita pela primeira vez pelos médicos Milton Riley e Richard Lawrence Day. Sem o aviso de perigo que a dor proporciona às pessoas comuns, a maioria dos doentes com a síndrome de Riley-Day tende a morrer jovem, antes dos 30 anos, por causa de ferimentos.
Síndrome de Moebius
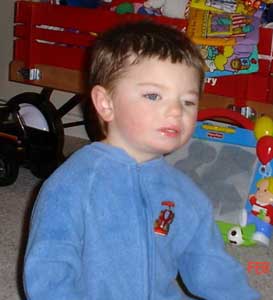
Frequência: Ao redor de 80 casos documentados na Espanha, 200 na Inglaterra e 5 na Argentina.
Causa: Desconhecida. Nem sequer sabe-se se são os nervos, o tronco do encéfalo ou os músculos que são afetados na origem da doença. Existem muitas e variadas hipóteses mas sem provas que as validem. Não há ainda uma explicação científica para a ocorrência da síndrome, que pode estar associada a diferentes fatores. Porém há fortes indicios de que seu surgimento esteja relacionado ao Misoprostol, medicamento conhecido como Citotec, usado para tratamento de úlcera gástrica, porém vendido clandestinamente para uso em tentativas de aborto.
Descrição: Devido ao não desenvolvimento de nenhum nervo facial, as pessoas que nascem com esta síndrome carecem de expressão facial. Não podem sorrir, nem franzir a testa, etc. Também não podem mover lateralmente os olhos nem controlar a piscada dos olhos. Com freqüência são encontrados dormindo com os olhos abertos. Têm grandes dificuldades em soprar, engolir, falar e qualquer atividade na que estejam implicados os músculos da face. é um distúrbio neurológico extremamente raro. Decorre do desenvolvimento anormal dos nervos cranianos, possui como principal característica a perda total ou parcial dos movimentos dos músculos da face, responsáveis pelas expressões e motricidade ocular.
Tratamento: Essa sindrome não possui cura, seu tratamento tem como objetivo proporcionar maior qualidade de vida ao portador, inclui cirurgias corretivas (ortopedia e estrabismo), fisioterapia, fonoterapia e terapia ocupacional.
Hermafroditismo

Frequência: Ao redor de 500 casos documentados em todo mundo. Desconhece-se a frequência real na população.
Causa: A pessoa hermafrodita é uma quimera. Produz-se pela fusão de dois zigotos de sexos diferentes. Isto é, primeiro um espermatozóide fecundaría um óvulo e depois outro espermatozóide fecundaría um outro óvulo. Os zigotos formados estariam destinados a serem gêmeos, mas acabam fundindo-se e se tornando um único indivíduo que, geneticamente, é mulher e homem ao mesmo tempo. Desconhece-se por que se produz esta fusão.
Descrição: Os hermafroditas têm tanto tecido ovárico como testicular. Os genitais externos são ambíguos e possuem componentes de ambos sexos. As pessoas hermafroditas podem ter aparência tanto feminina como masculina. Chama-se hermafrodita (do nome do deus grego Hermafrodito, filho de Hermes e de Afrodite – respectivamente representantes dos gêneros masculino e feminino) um ser ou animal que possui órgãos sexuais dos dois sexos, numa espécie dioica (ou seja, em que normalmente os sexos se encontram em indivíduos separados) podem aparecer indivíduos hermafroditas, mas geralmente por um processo teratológico, ou seja, por uma má formação embrionária.
Generalidades: Nas plantas verdes, a norma é a monoica, ou seja, cada indivíduo possuir os órgãos sexuais dos dois sexos.
Em muitas espécies de peixes, como as garoupas, verifica-se um tipo de hermafroditismo insuficiente, ou seja, os indivíduos possuem órgãos sexuais masculinos e femininos, mas apenas um dos tipos se encontra activo num determinado momento. Normalmente, o animal atinge a maturidade sexual com um determinado sexo e, no processo de crescimento, as gónadas convertem-se no outro sexo e tornam-se activas mais tarde. Nas espécies em que o sexo feminino é o primeiro a se tornar activo, diz-se que a espécie é protogínica. No caso inverso, diz-se protândrica.
Hermafroditismo humano: Existem três tipos de hermafroditismo humano: o hermafroditismo verdadeiro, o pseudo-hermafroditismo masculino e o pseudo-hermafroditismo feminino:
No hermafroditismo verdadeiro as crianças nascem com os dois órgãos sexuais bem formados, possuindo os oŕgãos sexuais internos e externos de ambos os sexos, incluindo ovários, útero, vagina, testículos e pênis. No hermafroditismo verdadeiro a maioria das pessoas são geneticamente do sexo feminino (cromossomos XX) e a formação dos órgãos sexuais masculinos é atribuída a causas ainda não totalmente conhecidas.
No pseudo-hermafroditismo masculino a criança nasce geneticamente como do sexo masculino (cromossomos XY) embora os órgãos sexuais externos não se desenvolvam completamente.
No pseudo-hermafroditismo feminino a criança nasce geneticamente como do sexo feminino (cromossomos XX) embora o clítoris desenvolva-se excessivamente adquirindo um formato semelhante a um pênis. Atribui-se uma suposta causa não genética para o pseudo-hermafroditismo feminino aos efeitos dos medicamentos utilizados no tratamento da hiperplasia congênita das supra-renais (HCSR) por deficiência da 21-Hidroxilase, uma doença genética que necessita de tratamento permanente e que em alguns casos é não é interrompido por gestantes que não sabem estar grávidas.
Uma teoria genética recente busca explicar várias anomalias sexuais do hermafrotitismo humano com sequências palíndromos presentes no cromossomo Y. Segundo essa teoria as sequências palíndromos presentes no cromossomo Y, que supostamente protegeriam esse cromossomo de mutações genéticas, poderiam ocasionalmente se esticar e formar uma atração fatal com o palíndromo similar de seu vizinho, alterando o tamanho e/ou deslocando o centrômero do gene: os cromossomos gerados nessas divisões celulares teriam comprimentos variáveis, curtos e longos, com centrômeros deslocados ora para o centro, ora para as extremidades. Nessa teoria, os pacientes nos quais a distância entre os dois centrômeros do Y é curta, seriam homens, ao passo que quanto maior a distância entre os centrômeros, maior a tendência de que os pacientes sejam anatomicamente feminilizados. Essa pesquisa incluiu alguns pacientes do sexo masculino (cromossomos XY) portadores da síndrome de Turner, uma condição só então conhecida em mulheres que nascem com um único cromossomo X (cromossomos 45-XO).
Tratamento: No tratamento do hermafroditismo humano recorre-se muitas vezes a uma cirurgia para se definir o sexo definitivo. Segundo especialistas a maior dificuldade está em se definir o momento correto da cirurgia. De todo o modo a opinião crescente é de que a pessoa hermafrodita possa escolher por si mesma se ela deseja a cirurgia e, nesse caso, qual o sexo desejado.
Fibrodisplasia ossificante progressiva
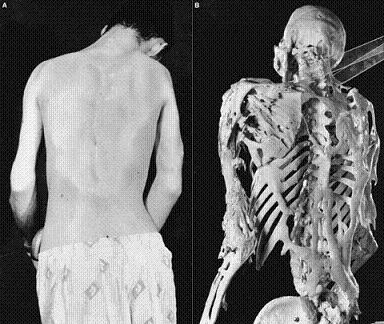
Frequência: 200-300 casos documentados em todo mundo. Os poucos conhecimentos que se tem da doença, muitas vezes, impossibilita o diagnóstico. Estima-se que surja um caso para cada dois milhões de nascimentos.
Causa: Desconhecida. É uma doença de herança autossômica dominante. Pensa-se que estão implicados vários genes encarregados de sintetizar fatores de crescimento ósseo. A formação de tecido ósseo no interior de músculos, tendões e ligamentos, causa de forma progressiva, a imobilização do corpo. É caracterizada por má-formação congênita do hálux (dedo grande dos pés malformados ao nascimento) e pelo desenvolvimento de ossos "extras" em locais anormais. Estes ossos surgem progressivamente e formam “pontes” entre as articulações, tornando os movimentos impossíveis.
Descrição: Nesta doença dão-se episódios repetidos de inflamação dos tecidos macios e o desenvolvimento de tumores subcutâneos e nos músculos. Estas lesões provocam a formação de osso em lugares onde nunca deveria existir osso, como ligamentos, músculos, tendões, articulações… Os traumatismos também desencadeiam e fazem avançar a ossificação dos tecidos macios. Progressivamente, o indivíduo irá perdendo cada vez mais a mobilidade até que, por impossibilidade de movimento da musculatura encarregada da respiração, morre por asfixia. A Fibrodisplasia Ossificante Progressiva (conhecido por FOP; termo médico: Fibrodysplasia Ossificans Progressiva) é uma doença genética rara que causa a formação de ossos no interior dos músculos, tendões, ligamentos e outros tecidos conectivos. Pontes de ossos "extra" se desenvolvem através das articulações (juntas do corpo) restringindo progressivamente os movimentos. Na FOP, o corpo não somente produz muitos ossos, mas um todo um esqueleto "extra" é formado, envolvendo o corpo e prendendo a pessoa em uma prisão de ossos. As crianças com FOP desenvolvem inchaços dolorosos pelo corpo, semelhantes a tumores, que podem crescer, mudar de posição e desaparecer, porém estes inchaços costumam deixar no seu lugar um osso e vão progressivamente imobilizando o corpo da criança num “segundo esqueleto”. O progresso da FOP pode ser espontâneo, ou ser acelerado por traumas (quedas, cirurgias, biópsias).
PARTE 2